Quick start with Physcraper¶
Updating a tree from Open Tree of Life¶
The Open Tree of Life data store, Phylesystem, contains more than 4,500 phylogenetic trees from published studies. The tips in these trees are mapped to a unified taxonomy, which makes these data searchable in a phylogenetically explicit way. This is a great place to start of finding existing estimates of phylogenetic relationships, and assessing regions of the tree of life which are lacking available phylogenetic estimates. There is a lot of sequence data available that has never been incorporated into any phylogenetic estimates.
Find a starting tree with your taxon of interest¶
For this example we will use a tree that is already in the Open Tree of Life database. You can find more details about finding a tree to update at the start section of this documentation.
To find trees containing your taxon of interest (e.g. ‘Malvaceae’) on OpenTree use:
$ find_trees.py --taxon_name "Malvaceae"
This prints a bunch of studies out to the screen. We will need an alignment to update (which OpenTree does not store), so let’s just look at trees that have data stored in TreeBASE.
$ find_trees.py --taxon_name "Malvaceae" --treebase
There are a bunch of options!
Lets update the Wilkie et al. 2006 (https://doi.org/10.1600/036364406775971714) study. You can view the study on the OpenTree database at Wilkie, 2006
While this study was focused on the family Sterculiacea, phylogenetic inference have suggested that this taxon is not monophyletic, as you can see on its OpenTree homepage)
Let’s take a look at how recent molecular data affect our inferences of relationships, and if there is sequence data for taxa that don’t have any phylogenetic information available in the tree.
Run the auto-update¶
The script physcraper_run.py wraps together linking the tree and alignment, blasting, aligning sequences, and inferring an updated tree.
Detailed explanation of the inputs needed can be found in the Run section of this documentation.
The BLAST search part of updating trees takes a long time. For example, this analysis took around 12 hours! We recommend running it on a cluster or other remote computing option.
$ physcraper_run.py -s pg_55 -t tree5864 -tb -r -o pg_55
The -r flag repeats the search on new sequences until no additional sequences are found.
We have put example outputs from this command in docs/examples/pg_55, so that you can explore the outputs without waiting for the searches to complete.
Updating your own tree and alignment¶
You can upload your own tree to OpenTree to update it, and that way it will be included in the OpenTree synthetic tree! See Submitting-phylogenies-to-Open-Tree-of-Life for more info on this.
If you aren’t ready to share your tree publicly, you can update it without posting it to OpenTree.
You need an alignment (single locus) and a tree. Note that the taxon labels in these two files should be the same.
You also need a file linking the labels in your tree and alignment to broader taxonomy. This can be easily generated via OpenTree’s Bulk Taxonomic Name Resolution Service (bulk TNRS).
Automatically mapping names to taxa¶
You can automatically map your tip names to unique taxonomic identifiers using OpenTree’s bulk Taxonomic Name Resolution Service (TNRS) tool. This can be done using R or Python programming languages, and the graphical user interface (GUI) version is available at https://tree.opentreeoflife.org/curator/tnrs/.
⚠ This is a brand new beta-version of this functionality, so some parts might be a bit finicky.
The first step is to save your tip taxon labels in a “.txt” file. There is an example file in the Physcraper folder at docs/examples/example_tiplabels.txt
Then, got to https://tree.opentreeoflife.org/curator/tnrs/ and click on add names, and upload the names file.

In the mapping options section, you can select a taxonomic group to narrow down the possibilities and speed up mapping. You can use regular expressions to replace or remove parts of labels for mapping.
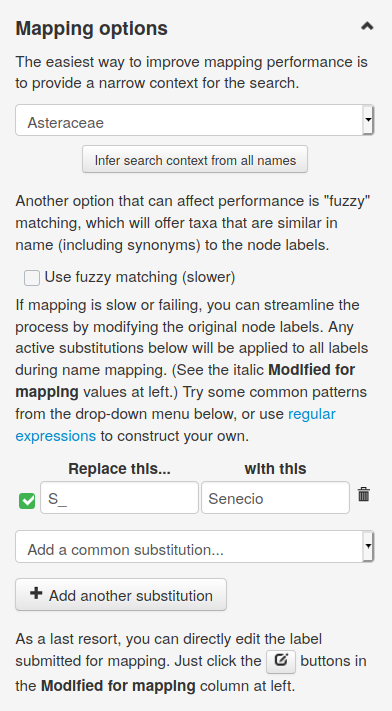
Click on Map selected names
Exact matches will show up in green, and can be accepted by clicking accept exact matches.
Some taxa may show several suggested names. Click through to the taxonomy, and select the one that you think is correct based on the phylogenetic context.

Once you have accepted names for each of the taxa, click save nameset.
⚠ Make sure your mappings were saved! If you don’t click on ``accept matches``, they don’t download.
Download your results to your laptop.
Extract the files.
Take a look at the human readable version at output/main.csv. You will see that this file also links to NCBI and GBIF identifiers for your taxa!
output/main.json contains the same data in a more computer readable format.
By passing in the main.json file, Physcraper can link your sequences to their correct taxonomic context.
Run the auto-update on your tree¶
Example run on local files using test data:
physcraper_run.py -tf tests/data/tiny_test_example/test.tre -tfs newick -a tests/data/tiny_test_example/test.fas --taxon_info tests/data/tiny_test_example/main.json -as fasta -o owndata